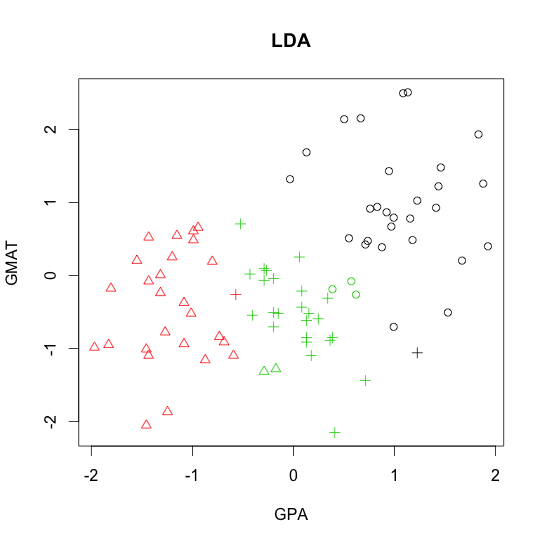
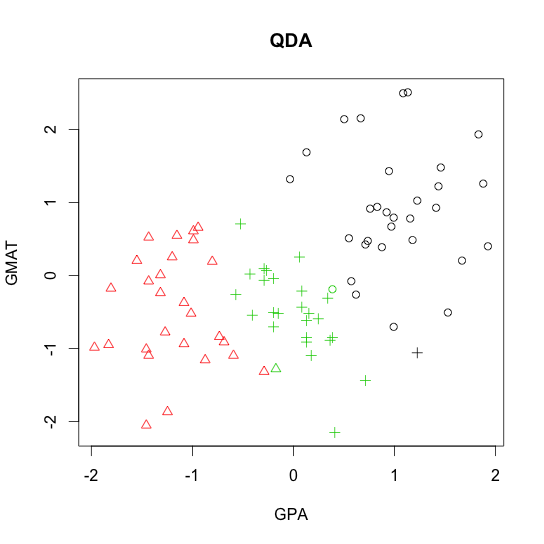
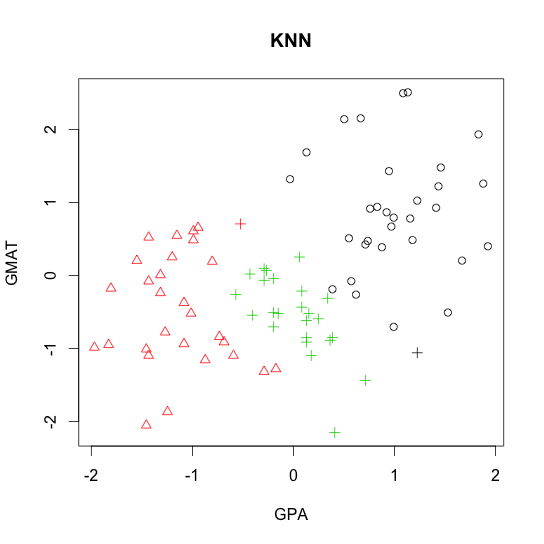
R code demo of
- Linear discriminant analysis (LDA)
- Quadratic discriminant analysis (QDA)
- k-nearest neighbor (KNN)
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 | |
1 2 3 4 5 6 7 8 | |
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 | |
R code demo of
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 | |
1 2 3 4 5 6 7 8 | |
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 | |
Bridge regression is a broad class of penalized regression, and can be used in high-dimensional regression problems.
It includes the ridge (q=2) and lasso (q =1) as special cases.
More technical details can be found here. Below R code demonstrates:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148 149 150 151 152 153 154 155 156 157 | |

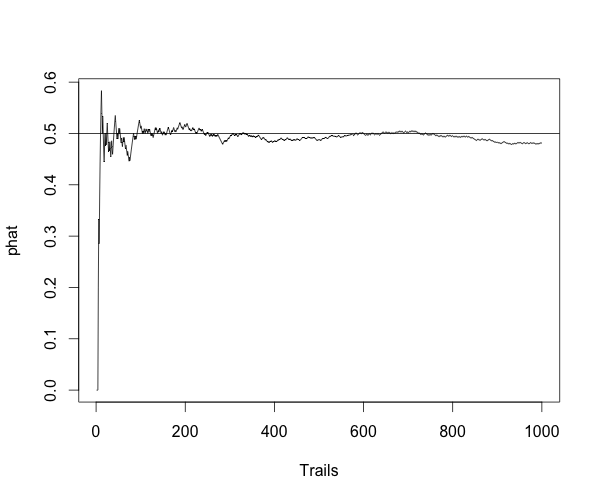
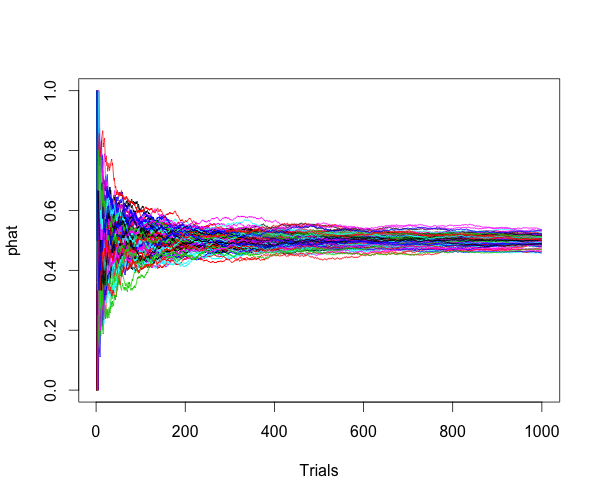
This is a simple post showing the basic knowledge of statistics, the consistency.
For Bernoulli distribution, $ Y \sim B(n,p) $, $ \hat{p}=Y/n $ is a consistent estimator of $ p $, because:
for any positive number $ \epsilon $.
Here is the simulation to show the estimator is consitent.
1 2 3 4 5 6 7 8 9 | |
1 2 3 4 5 6 7 8 9 10 11 12 13 14 | |
The procedure of permutation test for PCA is as follows:
For each replicate,
Individually permute each column of the data matrix.
Conduct the PCA and find the proportion of variance explained by each of the components 1 to s. Store this information.
Repeat 1 and 2 R times.
At the end of this we will have a matrix with R rows and s columns that contains the proportion of variance explained by each component for each replicate.
Finally, compare the observed values from the original data to the set of values from the permutations in order to determine the approximate p-value.
The R code:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 | |
The result:
$pve
Comp.1 Comp.2 Comp.3 Comp.4 Comp.5 Comp.6 Comp.7 Comp.8
0.23129378 0.14864525 0.11552865 0.06741744 0.06274641 0.05858431 0.05033795 0.04484122
Comp.9 Comp.10
0.03873311 0.03431297
$pval
[1] 0.000 0.000 0.000 1.000 1.000 0.996 1.000 1.000 1.000 1.000
Demo of SVM
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 | |
Cross validation for linear model and the bootstrap confidence interval for coefficients
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 | |
This post shows how to estimate gamma distribution parameters using (a) moment of estimation (MME) and (b) maximum likelihood estimate (MLE).
The probability density function of Gamma distribution is
The MME:
We can calculate the MLE of $ \alpha $ using the Newton-Raphson method.
For $ k =1,2,…,$
where
Use the MME for the initial value of $ \alpha^{(0)} $, and stop the approximation when $ \vert \hat{\alpha}^{(k)}-\hat{\alpha}^{(k-1)} \vert < 0.0000001 $. The MLE of $ \beta $ can be found by $ \hat{\beta} = \bar{X} / \hat{\alpha} $.
Below is the R code.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 | |
只收集一些相关资料,不评论。 http://www.sda.gov.cn/WS01/CL0051/102239.html 2014年6月30日,国家食品药品监督管理总局经审查,批准了BGISEQ-1000基因测序仪、BGISEQ-100基因测序仪和胎儿染色体非整倍体(T21、T18、T13)检测试剂盒(联合探针锚定连接测序法)、胎儿染色体非整倍体(T21、T18、T13)检测试剂盒(半导体测序法)医疗器械注册。这是国家食品药品监督管理总局首次批准注册的第二代基因测序诊断产品。
该批产品可通过对孕周12周以上的高危孕妇外周血血浆中的游离基因片段进行基因测序,对胎儿染色体非整倍体疾病21-三体综合征、18-三体综合征和13-三体综合征进行无创产前检查和辅助诊断。
http://www.knowgene.com/question/677 BGISEQ-1000基因测序仪基于Complete Genomics平台,配套的试剂盒为胎儿染色体非整倍体(T21、T18、T13)检测试剂盒(联合探针锚定连接测序法)。CG平台的特点是通量高,但周期较长,因此BGISEQ-1000应该主要会应用于全国范围内的样品,集中测序分析;
BGISEQ-100基因测序仪基于Ion Torrent平台,配套的试剂盒为胎儿染色体非整倍体(T21、T18、T13)检测试剂盒(半导体测序法)。Ion Torrent平台的特点是测序周期短,可灵活部署,BGISEQ-100有很大可能会被部署到有一定业务量的大中型医院,就地采样、测序、分析并出具报告.
The free online course Statistical Learning will start tomorrow. It will be taught by Rob Tibshirani and Trevor Hastie. It is an excellent opportunity to learn directly from the two famous professors in the field and the authors of two great textbooks on statistical learning, An Introduction to Statistical Learning, with Applications in R and The Elements of Statistical Learning.